
I'm trying to do density guided homology modeling using Rosetta CM. I have the following two issues:
1) I generated two threaded models, via setup_RosettaCM.py and manual threading. They are drastically different from one another.
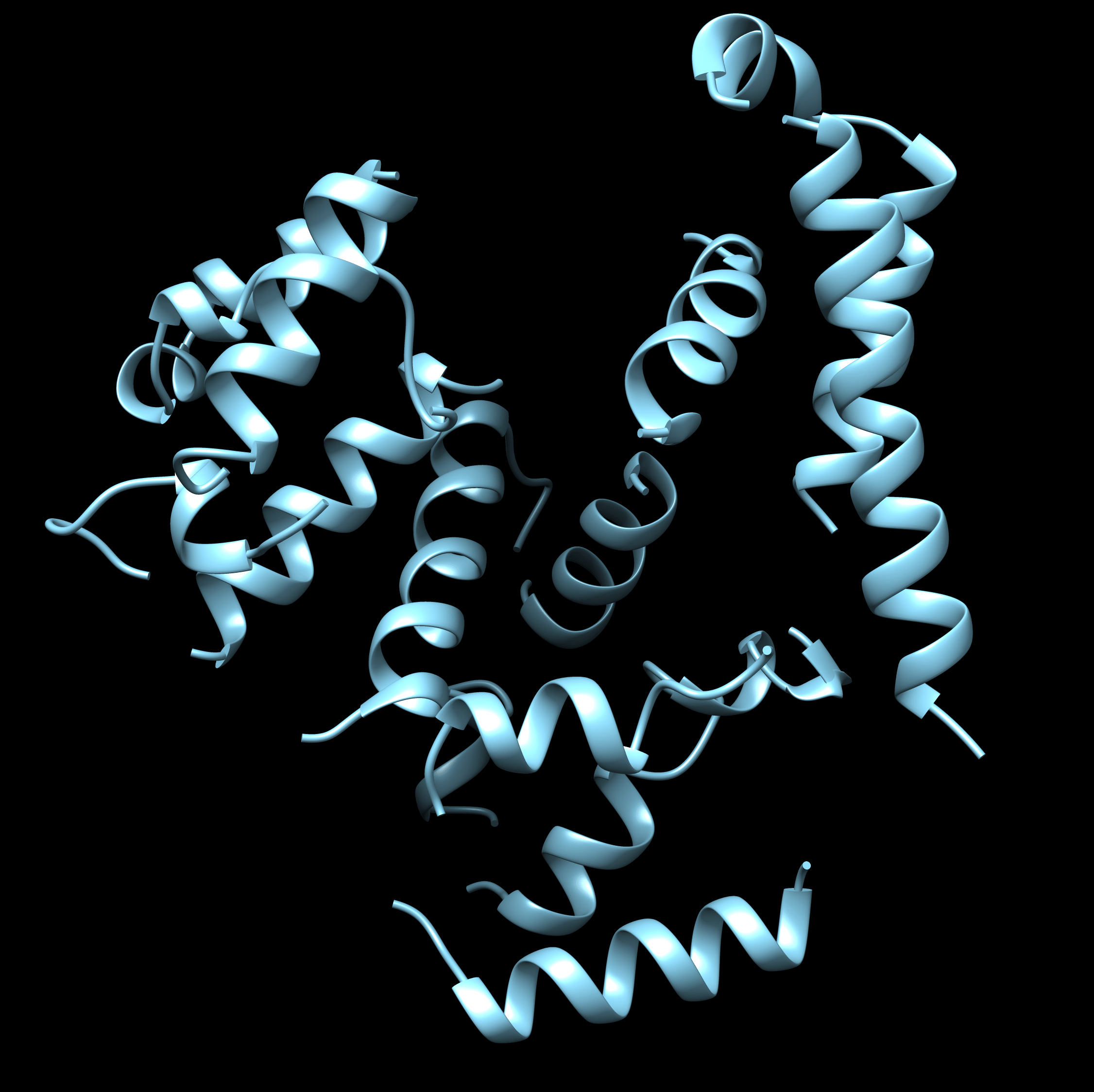
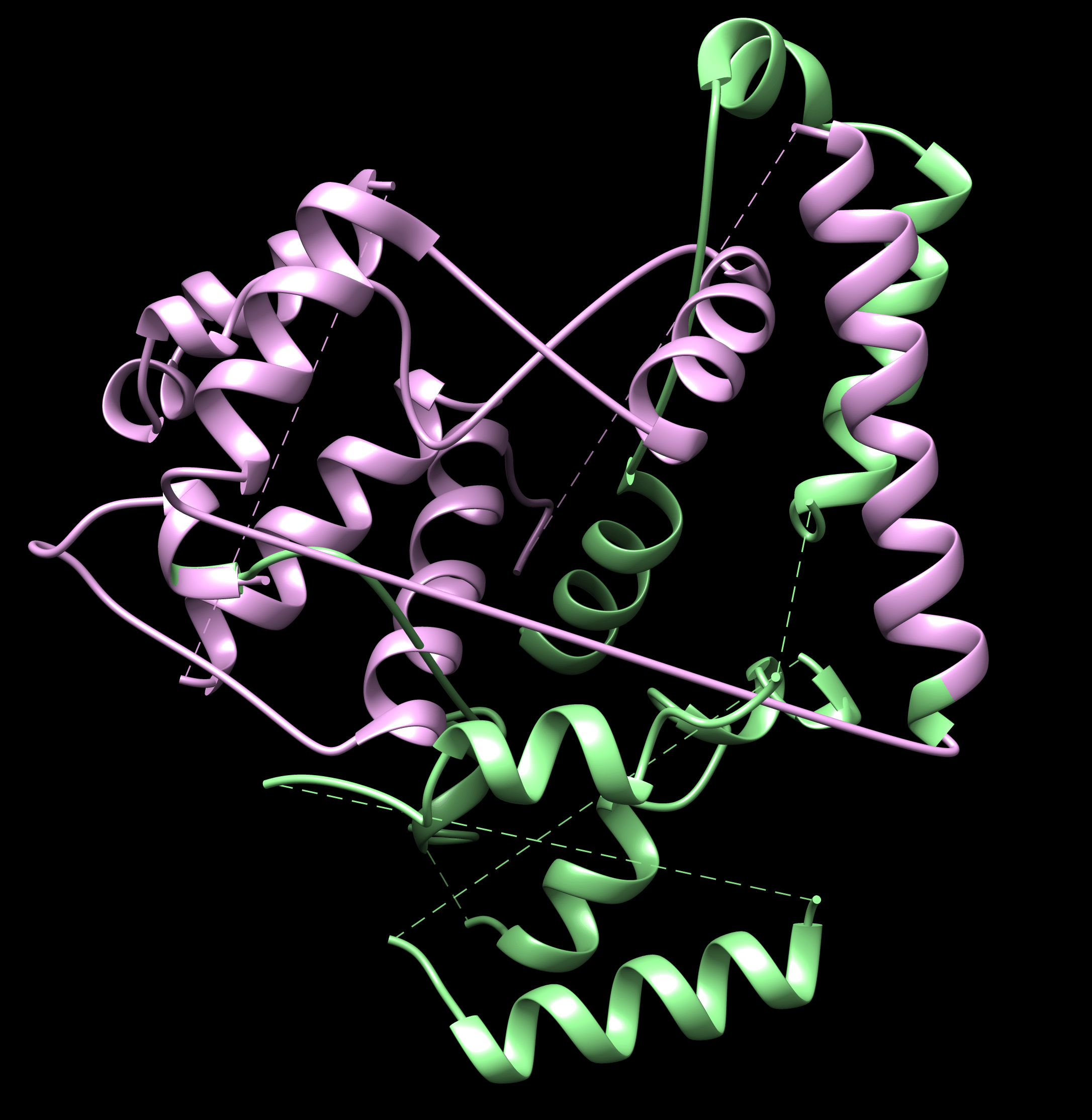
2) I verified that the template helices match those in the ground truth structure. I would expect from the threaded model to connect the helices in the template correctly according to the alignment between the template and ground truth sequence. As shown in the screenshot, both threaded models connect the backbone incorrectly, and I guess this is what's causing divergence in the rosetta_cm generated structures.
I attached screenshots for the template, template aligned with the auto threaded model and the manual threaded model.
| Attachment | Size |
|---|---|
| 352.15 KB | |
| 177.84 KB | |
| 375.68 KB |
{kind=link}
{kind=link}
{kind=link}
I updated the screenshots, I would appreciate your prompt input. In this google drive folder, you can find the following:
1) template.
2) manual threaded model.
3) auto threaded model.
4) template sequence.
5) ground_truth_sequence.
6) grishin alignment file.
https://drive.google.com/drive/folders/1cvLO27_3Spd5jF-TTudb8VUW8L8AdU4z?usp=sharing
I just realized, some of the fragments are placed incorrectly, this is likely why the threaded model is a bit messed up. In real de novo situation, where there is no homologue, how to be able to detect this? Still, why am I getting two diffeent models from auto and manual threading?
I'm not 100% on what's going on here, but I'm guessing it's that the automated process is finding a different alignment than the manual one is.
The quality of threading is totally due to how good your alignment is. If you have a single protein you care about, it's totally wort the time to spend several days tweaking and optimizing the alignment. That's why I often recommend a more manual proceedure. The automated script is good if you're dealing with a large number of proteins (e.g. in a CASP competition), but if there's just one protein or one protein family you're interested in, it's often worth manually doing the alignment and checking that the threaded structure makes sense. (Purely sequence-based methods can do odd things like break the alignment in the middle of a helix, or smash two sides of a gap the wrong way around.)
I can't be certain without seeing the Grishin file the automated protocol puts out, but I think what's happening here is the automated protocol isn't finding the same alignment your manual approach is, and it's messing it up. -- It's slightly curious, as this seems to be a relatively straightforward alignment, but the automated protocol is optimized for medium-low sequence identity cases, so those heuristics might be messing things up for your "easier" case.
All in all, if you're comfortable with doing things manually, I'd stick to that, and sanity-check your threaded alignments prior to going on to the next step.
Finally, I'm not sure what you mean by "where there is no homologue". RosettaCM is homology modeling, so it requires a homolog structure (or at the least, a structure with the same fold which you can thread onto). If you don't have that, attempting to use RosettaCM is pointless - you'll want to use a different protocol like AbRelax and the like, which don't use any template information. (Slight terminology clarification - typically we don't refer to the threaded structure as "fragments" - fragments are normally just sequence-based subsections of proteins from non-homologous proteins. The concept is that small sections of protein fold the same way, even if the overall topology is completely different. -- RosettaCM uses fragments to fill in gaps, but it primarily relies on the overall topologies generated by threading onto structurally similar proteins.)
Thanks for your feedback. Yeah I'm 100% sure now that the thgreading is messing my template, connecting wrong fragments together in a way that doesn't align with the sequence. Have you checked this folder here https://drive.google.com/drive/folders/1cvLO27_3Spd5jF-TTudb8VUW8L8AdU4z?usp=sharing for the grishin file?
I'm doing de novo first, so that's where my homologue or "template" comes from, that's what I meant by no homologue.
Do you have recommendation for a way of aligning the sequences and a strategy for checking on the alignment?
My recommended process for RosettaCM is to start with an automatic sequence alignment (e.g. from Clustal or Muscle or your favorite sequence alignment tool), and then start tweaking it. (I'd even run the alignment through multiple methods to see different possibilities.)
You can start with manual tweaks (e.g. "well, I know that *this* is a conserved cysteine, so those should be aligned, and here's the binding motif, so I probably should align those, too), but for the main part is likely to be an iterative process of taking a potential alignment, running the threading, then staring at the threaded structure. You can pretty easily see major issues (like threading putting a gap in the middle of a well-conserved helix), but you'll also want to check for "hidden" issues like insertions in places which probably can't accomodate them (you can note insertions by a skip in numbering), or too-short loops, or having helicies or sheets shifted (e.g. the hydrophobic/hydrophilic patterning doesn't match the structure you're threading over.)
Often it's a bit of a judgement call as to what the "best" alignment is, and you can use your system-specific knowledge to guide it. At a certain point you're going to want to run the prospective alignment through the whole modeling protocol, and look at the quality of the results. Keep in mind you can always model based on multiple different alignments, and then pick the best structure from the different alignments.
But from what I remember of your system, you have pretty good sequence identity between the templates and the target, so there probably isn't much to do with respect to fiddling with the alignment. You can just do the obvious alignment, then check the threading to make sure you're not messing things up totally. And if your threading is messed up with the obvious alignment, it may be that the template structure just isn't a good one to use in the CM protocol. (This is potentially relevant in your case, if you're using de novo predictions as possible templates for some of the domains.)
My recommended process for RosettaCM is to start with an automatic sequence alignment (e.g. from Clustal or Muscle or your favorite sequence alignment tool), and then start tweaking it. (I'd even run the alignment through multiple methods to see different possibilities.)
You can start with manual tweaks (e.g. "well, I know that *this* is a conserved cysteine, so those should be aligned, and here's the binding motif, so I probably should align those, too), but for the main part is likely to be an iterative process of taking a potential alignment, running the threading, then staring at the threaded structure. You can pretty easily see major issues (like threading putting a gap in the middle of a well-conserved helix), but you'll also want to check for "hidden" issues like insertions in places which probably can't accomodate them (you can note insertions by a skip in numbering), or too-short loops, or having helicies or sheets shifted (e.g. the hydrophobic/hydrophilic patterning doesn't match the structure you're threading over.)
Often it's a bit of a judgement call as to what the "best" alignment is, and you can use your system-specific knowledge to guide it. At a certain point you're going to want to run the prospective alignment through the whole modeling protocol, and look at the quality of the results. Keep in mind you can always model based on multiple different alignments, and then pick the best structure from the different alignments.
But from what I remember of your system, you have pretty good sequence identity between the templates and the target, so there probably isn't much to do with respect to fiddling with the alignment. You can just do the obvious alignment, then check the threading to make sure you're not messing things up totally. And if your threading is messed up with the obvious alignment, it may be that the template structure just isn't a good one to use in the CM protocol. (This is potentially relevant in your case, if you're using de novo predictions as possible templates for some of the domains.)