De novo structure prediction
KEYWORDS: STRUCTURE_PREDICTION GENERAL
Written by:
Sebastian Rämisch
raemisch@scripps.edu default.linuxclan
This tutorial will teach you how to predict protein structures starting with an amino acid sequence. You will predict the structure of bacteriophage T4 lysozyme using ab initio protein folding. At the end of the tutorial, the results will be compared to the native structure, which is available from the PDB.
1. Prepare your input files.
You will need:
- The sequence in fasta format
- 9mer fragment file
- 3mer fragment file
For this tutorial: the known native structure (pdb file)
-
Save your protein sequence to a file using FASTA format.
Here, a fasta file is provided for you.$ cat input_files/2LZMA.fasta >2LZMA ITKDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRCALINMVFQMGETGVAGFTNSLRMLQQKRWDEAAVNLAKSRWYNQTPNRAKRVITTFRTGTWDAYKNL -
Prepare fragment files. Those will contain short backbone fragments that will be randomly inserted at all positions during the simulation. Here, fragment files are provided:
input_files/aa2LZMA03_05.200_v1_3 (3mer fragments) input_files/aa2LZMA09_05.200_v1_3 (9mer fragments)
There are multiple ways to obtain those fragment files. You can generate them on your local machine. For that to work you need additional software installed. How to do that is explained in a separate tutorial for fragment picking. Alternatively, you can submit your fasta file to the Robetta server:
http://robetta.bakerlab.org/
(3.) Copy the reference structure (if there is one) into your input file directory. Here, the native structure file is provided, so you can compare the generated models to the known native structure.
input_files/2LZMA.pdb
2. Run the Rosetta AbinitioRelax application.
# For a production run on a cluster, run AbinitioRelax.mpi.linuxclangrelease
$> ../../../main/source/bin/AbinitioRelax.default.linuxclangrelease @input_files/options
Alternatively, run in the background:
$ nohup ../../../main/source/bin/ AbinitioRelax.default.linuxclangrelease @input_files/options > log &
NOTE: This will take 10-20 minutes per structure.
Expected output:
- S_00000001.pdb (the generated model)
- score.sc (shows the total score and individual terms)
- For an actual production run, 50,000 to 100,000 models need to be generated.
- You probably want to use the silent file options
3. Analyze your results
Details of how to analyze data and how to select which of the 50,000 models would be your prediction are explained in the Analysis tutorial.
Short walk through of initial steps
For de novo structure prediction, in general you will want to perform the following steps:
3.1. Plot score vs. rmsd
To see how confident you can be about the correctness of a prediction, you can plot score vs. rmsd for the top 5% or 10% of the models. Both total_score and rms are provided in the score file. This is only possible, if the native structure is known. In a real world case, you could use a homologous protein. However, if no related structure is known, you would use e.g. the lowest energy model (and rescore the top models with e.g. -native best_E.pdb).
- Find in which column total_score and rms is reported (here, column 25).
-
Then try:
$ sort -n -k2 score.sc | head -n 5000 | awk '{print $25 "\t" $2}' > score_rmsd.dat-> The new file (score_rmsd.dat) contains the scores from the top 5000 models.
-
Use your favourite ploting program and plot score_rmsd.dat as a scatter plot.
Here are examples of such plots:
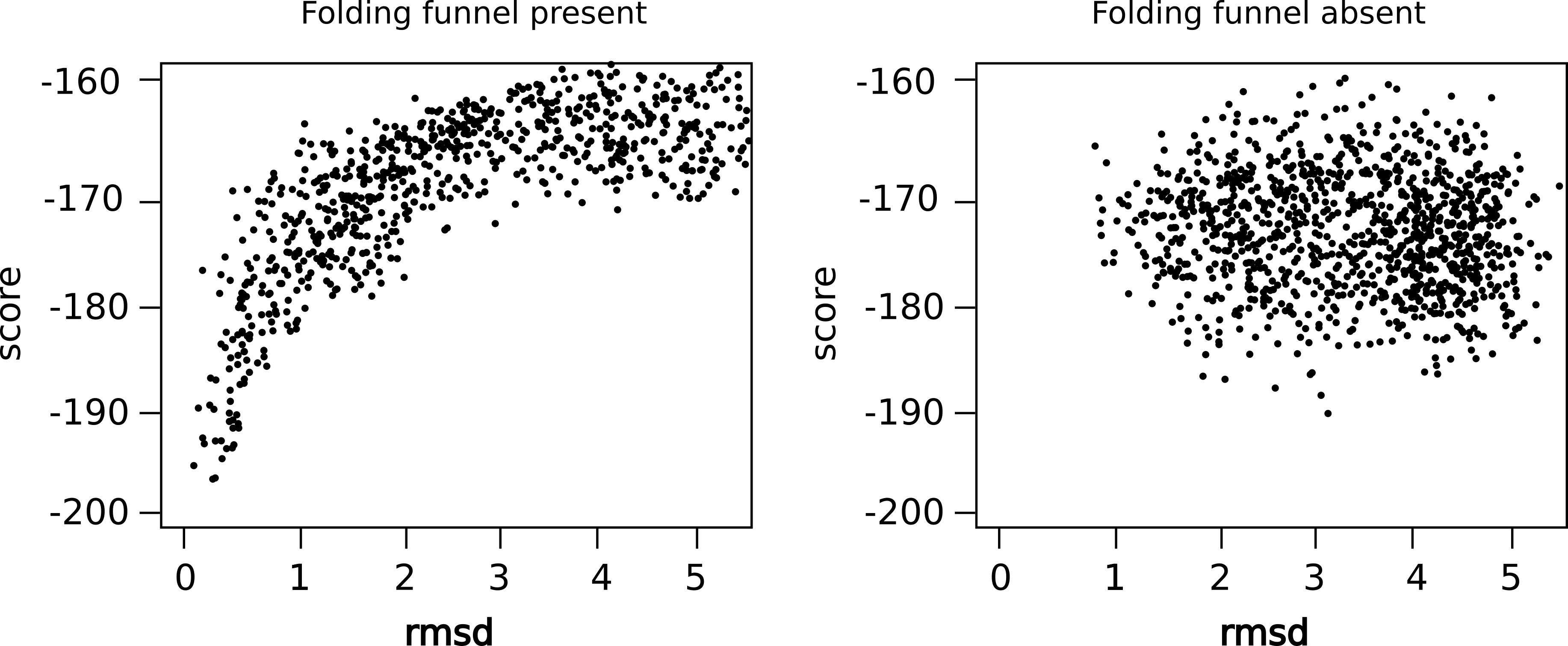
The left plot shows that the lowest energy models also have the lowest rmsd - the simulation converged. On the right hand side, however, there is no convergence. Many very different models have low energies. A prediction would be highly questionable.
The plot you will get, should converge towards an rmsd of approx. 5 Angstrom. You can see what a larger run will look like by plotting score_rmsd.dat provided in example_outputs directpry which is generated using the same steps described above..
3.2. Extract the best (by score) of the generated models.
5 Angstrom does not quite sound like Rosetta really solved the problem. The only way to find out is by looking at the best structure(s)
More instructions and helpful scripts are part of the Analysis tutorial (Analysis).
-
In the example_outputs directory, you will find a binary silent file from a full AbinitioRelax run (AbRelax.out) and score file (score.sc).
Run:$ cd output_files/ $ ls Identify the best 5 structures:
This will sort the score file by the second column (total_score) and print the first 5 lines to the screen. The last column gives you the name (tag) of a model.$ sort -n -k2 score.sc | head -l 5-
Extract those models from the silent file:
$ ../../../main/source/bin/extract_pdbs.default.linuxgccrelease -in:file:silent_struct_type binary -in:file:silent AbRelax.out -in:file:tags <e.g. S_0013 S_0780_1 ...>Now you can look at the best structure in a molecule viewer.
WARNING
A good Rosetta score does not allways mean that the structure is good. Inaccuracies in the energy function leave unfavourable features 'unpunished'. So, allways ask yourself:
- Does the result make sense?
3.3. Clustering analysis
This sorts the structures by similarity. (See Analysis)