Constraint File
Metadata
This document was edited by Colin Smith on 12/4/2008. Yi Liu created the initial page. Thanks Oliver and Firas for providing information. Last edited by Steven Lewis on 30 Aug 2016.
Overview
Rosetta constraints are additions to the scorefunction. (This corresponds to "restraints" in other programs.) They're used to score geometric and other features of the structure which may not be evaluated by standard scoreterms. For example, adding a scoring bias based on experimental knowledge.
Each constraint consists of two parts: A) what's being measured B) how that measured value is transformed into a scoring bonus/penalty. These two parts can be mixed and matched to derive the desired behavior.
In order for constraints to be correctly recognized by Rosetta, two things must occur. First, the constraints themselves must be applied to the pose (structure). How this is done is somewhat protocol dependent, but most often takes the form of an option or parameter which specifies which file contains the constraint specification. (The format of this file is described below.) For example, by adding it in your xml script using the ConstraintSetMover.
The second requirement is that the scorefunction being used needs to have a non-zero weight for the appropriate constraint scoreterm. The particular scoreterm depends on the type of constraint being used. The value of the penalty/bonus consists of the sum of the raw constraint scores (from the measured value and the specified transforming function of all the constraints) multiplied by the weight of the appropriate score term in the score function. Many protocols which use constraints will turn the constraint weights on for you, but others will require you to specify a scorefunction weights file which has non-zero constraint terms.
File Formats
Constraints can be specified in a line-based constraint file formatted like so:
Constraint_Type1 Constraint_Def1
Constraint_Type2 Constraint_Def2
...Generally speaking, the Constraint_Type will contain a type, defining what sort of value to be constrained (distance, angle, dihedral, etc), and a series of atom and/or residue labels defining a specific quality to be constrained. Residue numbers are assumed to be in Rosetta numbering (from 1, no gaps), not PDB numbering. If you want PDB numbering, pass the chain letter immediately after the residue number (no spaces): residue 30 of chain A would be "30A". You cannot pass insertion codes through this mechanism; you'll need the renumbered pose. Not all constraint types can take PDB numbering.
The Constraint_Def will define function by which the constraint is constrained, to answer the question: what should the score of the constraint be when the constrained value has a deviation of X units?
Constraint Types
Constraint types are all implemented as subclasses of the core::scoring::constraints::Constraint class.
Single constraints
Single constraints restrain the value of a single metric
-
AtomPair:
AtomPair Atom1_Name Atom1_ResNum Atom2_Name Atom2_ResNum Func_Type Func_Defscore term: atom_pair_constraint
- Constrains a distance between Atom1 and Atom2. AtomPairConstraint is compatible with PDB numbering.
-
NamedAtomPair:
NamedAtomPair Atom1_Name Atom1_ResNum Atom2_Name Atom2_ResNum Func_Defscore term: atom_pair_constraint
- Constrains a distance between Atom1 and Atom2.
- Atoms in NamedAtomPairConstraints are stored as names rather than numbers, as with AtomPairConstraint. Therefore, if atom numbers change while constraints are in the pose, the NamedAngleConstraint will still constraint the correct atoms, while the AngleConstraint may constrain unintended atoms. There is a small tradeoff in computational efficiency versus AngleConstraint. If you know your atom numbers will not change while the constraint is in the pose, AngleConstraint is a better option, but if they might change, NamedAngleConstraint will ensure that the correct atoms are constrained.
-
Angle:
Angle Atom1_Name Atom1_ResNum Atom2_Name Atom2_ResNum Atom3_Name Atom3_ResNum Func_Type Func_Defscore term: angle_constraint
- Angle between Atom2->Atom1 vector and Atom2->Atom3 vector; the angle (passed as a value to the Func) appears to be measured in radians
- NOTE: AngleConstraint uses atom numbers to internally track the constrained atoms for efficiency. If the atom numbers change while the AngleConstraint is in the pose, the constraint could be applied to the wrong atoms. If you know your atom numbers will not change while the constraint is in the pose, AngleConstraint is the best option, but if they might change (e.g. by design or changing residue type set), NamedAngleConstraint will ensure that the correct atoms are constrained.
-
NamedAngle:
NamedAngle Atom1_Name Atom1_ResNum Atom2_Name Atom2_ResNum Atom3_Name Atom3_ResNum Func_Type Func_Defscore term: angle_constraint
- Angle between Atom2->Atom1 vector and Atom2->Atom3 vector; the angle (passed as a value to the Func) appears to be measured in radians
- Atoms in NamedAngleConstraints are stored as names rather than numbers, as with AngleConstraint. Therefore, if atom numbers change while constraints are in the pose, the NamedAngleConstraint will still constraint the correct atoms, while the AngleConstraint may constrain unintended atoms. There is a small tradeoff in computational efficiency versus AngleConstraint. If you know your atom numbers will not change while the constraint is in the pose, AngleConstraint is a better option, but if they might change, NamedAngleConstraint will ensure that the correct atoms are constrained.
-
Dihedral:
Dihedral Atom1_Name Atom1_ResNum Atom2_Name Atom2_ResNum Atom3_Name Atom3_ResNum Atom4_Name Atom4_ResNum Func_Type Func_Defscore term: dihedral_constraint
- Dihedral angle of Atom1->Atom2->Atom3->Atom4. Dihedral is measured in radians on -pi -> pi.
-
DihedralPair:
DihedralPair Atom1_Name Atom1_ResNum Atom2_Name Atom2_ResNum Atom3_Name Atom3_ResNum Atom4_Name Atom4_ResNum Atom5_Name Atom5_ResNum Atom6_Name Atom6_ResNum Atom7_Name Atom7_ResNum Atom8_Name Atom8_ResNum Func_Type Func_Defscore term: dihedral_constraint
- Constrains that the dihedral angles defined by Atom1->Atom2->Atom3->Atom4 and Atom5->Atom6->Atom7->Atom8 be identical.
-
CoordinateConstraint:
CoordinateConstraint Atom1_Name Atom1_ResNum[Atom1_ChainID] Atom2_Name Atom2_ResNum[Atom2_ChainID] Atom1_target_X_coordinate Atom1_target_Y_coordinate Atom1_target_Z_coordinate Func_Type Func_Defscore term: coordinate_constraint
- Constrain Atom1 to the XYZ position listed. Atom2 is used as a reference atom to determine when Atom1 has moved (so that Rosetta knows when to rescore it) - pick at atom that Atom1 will move relative to, the root atom of the FoldTree being a good choice. CoordinateConstraint is compatible with PDB numbering.
- Atom_ResNum[Atom_ChainID] indicates a number with an optional letter together as a single token
-
LocalCoordinateConstraint:
LocalCoordinateConstraint Atom1_Name Atom1_ResNum Atom2_Name Atom3_Name Atom4_Name Atom234_ResNum Atom1_target_X_coordinate Atom1_target_Y_coordinate Atom1_target_Z_coordinate Func_Type Func_Defscore term: coordinate_constraint
- Constrain Atom1 to the XYZ position listed, relative to the coordinate frame defined by atoms 2/3/4 instead of the origin. LocalCoordinateConstraint is compatible with PDB numbering.
-
AmbiguousNMRDistance:
AmbiguousNMRDistance Atom1_Name Atom1_ResNum Atom2_Name Atom2_ResNum Func_Type Func_Defscore term: atom_pair_constraint
- Distance between Atom1 and Atom2. The difference from AtomPairConstraint is that atom names are specially parsed to detect ambiguous hydrogens, which are either experimentally ambiguous or rotationally identical (like methyl hydrogens). The constraint applies to any hydrogens equivalent to the named hydrogen. The logic for determining which hydrogens are which is in src/core/scoring/constraints/AmbiguousNMRDistanceConstraints.cc:parse_NMR_name.
-
SiteConstraint:
SiteConstraint Atom1_Name Atom1_ResNum Opposing_chain Func_Type Func_Defscore term: atom_pair_constraint
- Constraint that a residue interacts with some other chain. SiteConstraints are a set of ambiguous atom-pair constraints that evaluate whether a residue interacts with some other chain or region - roughly, that it is (or is not) in a binding site. More specifically, if we have a SiteConstraint on a particular residue, that SiteConstraint consists of a set of distance constraints on the C-alpha from that residue to the C-alpha of all other residues in a set, typically the set being specific residues on another chain or chains. After each constraint is evaluated, only the constraint giving the lowest score is used as the SiteConstraint energy for that residue. The atom and resnum identify which atom is being checked for interactions with the opposing chain. Notice that "Constraint" is irregularly in its tag. Regions for the SiteConstraint other than the entire other chain cannot currently be set via constraint file.
-
SiteConstraintResidues:
SiteConstraintResidues Atom1_ResNum Atom1_Name Res2 Res3 Func_Type Func_Defscore term: atom_pair_constraint
- Constraint that a residue interacts with at least one of two other residues. The atom and resnum identify which atom is being checked for interactions with the CA atoms of the other residues.
-
MinResidueAtomicDistance:
MinResidueAtomicDistance res1 res2 distscore term: atom_pair_constraint
- Constraint that at least one heavyatom from res1 falls within dist of at least one heavyatom from res2. Implemented via AmbiguousConstraint (see below); a FADE function is used for each atom pair.
-
BigBin:
BigBin res_number bin_char sdevscore term: dihedral_constraint
- Specify wide bins for particular residues by letter. 'O' requires a cis-like omega angle (-10 to 10 degrees); 'G' requires positive phi and psi on [ -100, 100 ]; 'E' requires positive phi and psi on [ 100, -90 ]; 'A' requires negative phi and psi on [ -50, 30 ]; 'B' requires negative phi and psi on [ 100, 175 ].
Nested constraints
Nested constraints take as their parameters one or more other constraints, and allow optimization across multiple constraints. Typically in constraint files these are listed across multiple lines, with the name of the constraint opening the block of sub-constraints, and a line starting with "END" or "End" ending the block. In general, the scoretypes used by the nested constraints depends on which sub-constraints are used (this can normally be mixed).
-
MultiConstraint:
MultiConstraint Constraint_Type1 Constraint_Def1 [Constraint_Type2 Constraint_Def2 [...]] END- Sum of all specified constraints
-
AmbiguousConstraint:
AmbiguousConstraint Constraint_Type1 Constraint_Def1 [Constraint_Type2 Constraint_Def2 [...]] END- Lowest of all specified constraints
- Only the one with lowest energy is considered
-
KofNConstraint:
KofNConstraint k Constraint_Type1 Constraint_Def1 [Constraint_Type2 Constraint_Def2 [...]] END- Value is the sum of the "k" lowest energy constraints
Function Types
Functions are listed as "Func_Type Func_Def".
-
Specialized for angles:
-
CIRCULARHARMONIC x0 sd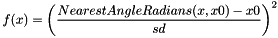
-
PERIODICBOUNDED period lb ub sd rswitch tag-
Note: Setting
rswitchto anything other than 0.5 will create a discontinuity in the derivative.rswitchandtagshould not be treated as optional.A BOUNDED constraint after mapping the measured value to the range -period/2 to +period/2. Useful for angle measures centered on zero.
-
-
OFFSETPERIODICBOUNDED offset period lb ub sd rswitch tagA BOUNDED constraint, where the measured value is (x - offset) mapped to the range -period/2 to +period/2. Useful for angle measures. (Note that lb and ub are interpreted after subtraction, so the true range of zero constraint is from lb+offfset to ub+offset.)
-
AMBERPERIODIC x0 n_period kAn AMBERPERIODIC function is a cosine function of the angle x, with a maximum at x0 and a periodicity of n_period. The amplitude is k, and the minimum value is 0:
- f(x) = k * (1 + cos( ( n_period * x ) - x0 ) )
-
CHARMMPERIODIC x0 n_period kA CHARMMPERIODIC function penalizes deviations from angle x0 by values from 0 to k, with n_period periods:
- f(x) = 0.5 * k * (1 - cos( n_period * ( x - x0 ) ) )
-
CIRCULARSIGMOIDAL xC m o1 o2A CIRCULARSIGMOIDAL function penalizes deviations x0 from angles o1 and/or o2 by values from 0 to 1, with n_period periods:
- f(x) = 1/(1+ M_E ^ (-m(x0-o1))) - 1/(1+M_E^(-m(x0-o2)))
-
CIRCULARSPLINE weight [36 energy values]A CIRCULARSPLINE function sets up a periodic cubic spline trained on the provided energy values, which represent the centers of thirty-six 10 degree bins
-
-
HARMONIC x0 sd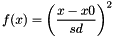
-
FLAT_HARMONIC x0 sd tolZero in the range of
x0 - toltox0 + tol. Harmonic with width parameter sd outside that range. Basically, a HARMONIC potential (see above) split at x0 with a 2*tol length region of zero inserted. -
BOUNDED lb ub sd rswitch tag- Note: Setting
rswitchto anything other than 0.5 will create a discontinuity in the derivative. (Iftagis not numeric, thenrswitchmay be omitted and will default to 0.5.)tagis NOT optional.

- Note: Setting
-
GAUSSIANFUNC mean sd tag WEIGHT weight- Note:
tagis NOT optional, as for BoundFunc/BOUNDED. Iftag= NOLOG, it triggers some undocumented behavior involving a logarithm of some sort. - Note: The string "WEIGHT" followed by a value is optional at the end. If provided, it scales the gaussian function by a constant multiplier. (For example, WEIGHT 2.5 scales the function by a factor of 2.5).
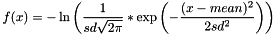
- Note:
-
SOGFUNC n_funcs [mean1 sdev1 weight1 [mean2 sdev2 weight2 [...]]]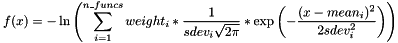
-
MIXTUREFUNC anchor gaussian_param exp_param mixture_param bg_mean bg_sd- parameters: representing the mean of h(r), representing the standard deviation of h(r)
-
CONSTANTFUNC return_val
-
IDENTITY- f(x) = x
-
SCALARWEIGHTEDFUNC weight Func_Type Func_Def
-
SUMFUNC n_funcs Func_Type1 Func_Def1 [Func_Type2 Func_Def2 [...]]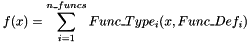
-
SPLINE description histogram_file_path experimental_value weight bin_sizeor, if the option
-constraints:epr_distanceis set:SPLINE description experimental_value weight bin_sizeor, if one wishes to provide a spline definition in a single line in a constraints file:
SPLINE description NONE experimental_value weight bin_size x_axis <val1> ... <valN> y_axis <val1> ... <valN>In the first form, this function reads in a histogram file and creates a cubic spline over it using the Rosetta SplineGenerator. The full path to the file must be specified. The basic form of the histogram file is a TAB SEPARATED file of the following format:
x_axis -1.750 -1.250 -0.750 -0.250 0.250 0.750 1.250 1.750 y_axis 0.000 -0.500 -1.000 -2.000 -1.500 -0.500 -0.250 0.000The values in the
x_axisline must be in ascending order, and it is assumed that the values are for the center of the histogram bin and that all bin widths are the same as that specified in the constraint file. (In practice, the bin_size setting is only used for the bins on the end.) It's assumed that the values return to the baseline at the edge of the given x_axis range, and that all y_axis values outside the range are zero.If the
descriptionparameter isEPR_DISTANCE, then the functional transformation of the measurement x isweight * S(experimental_value - x), otherwise the functional transformation isweight * S(x), andexperimental_valueis ignored.In the second form, if the
-constraints:epr_distanceoption is given on the command line, then thehistogram_file_pathshould be omitted, and the RosettaEPR knowledge-based potential will be read from a file in the Rosetta database. (See Hirst et al. (2011) J. Struct Biol. 173:506 and Alexander et al. (2013) PLoS One e72851 for details on this potential.) See example below for using with EPR knowledge-based potential.In the third form, if the
-constraints:epr_distanceoption is not used butNONEis substituted for thehistogram_file_path, then the spline definition must be provided on the same line. It must includex_axisfollowed by a series of values, andy_axisfollowed by a series of values. Optionally, it may includelb_function <cutoff> <slope> <intercept>to define a linear function for the curve past the lower bound, andub_function <cutoff> <slope> <intercept>to define a linear function past the upper bound. -
FADE lb ub d wd [ wo ]- This is meant to basically be a smoothed "square well" bonus of
wdbetween the boundarieslbandub. An optional offsetwo(default 0) can be added to the whole function; this is useful if you want to make the function be zero in the 'golden range' and then give a penalty elsewhere (e.g., specify wd of -20 and wo of +20). To make sure the function and its derivative are continuous, the function is connected by cubic splines in the boundary regions in slivers of width d, betweenlbtolb+dand betweenub-dtoub:
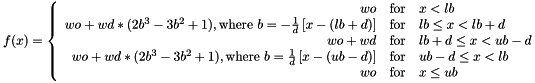
- This is meant to basically be a smoothed "square well" bonus of
-
SIGMOID x0 m- Two arguments; x0 is the center of the sigmoid func and m is its slope. It has hardcoded min/max of 0.5. The functional form is (1/(1+exp(-m*( x-x0 ))) - 0.5.
-
SQUARE_WELL x0 depth- f(x) = 0 for x >= x0
- f(x) = depth for x < x0
-
SQUARE_WELL2 x0 width depth [DEGREES]For angle measures. Parameters are presumed to be in radians unless optional DEGREES tag is present
- f(x) = 0 for (x0-width) < x < (x0+width)
- f(x) = depth for x <= (x0-width) and x >= (x0+width)
-
LINEAR_PENALTY x0 depth width slope- f(x) = depth for (x0-width) <= x <= (x0+width)
- f(x) = depth + slope * (abs(x-x0) - width) for abs(x-x0) > width
(Currently has a bug in minimization calculations.)
-
KARPLUS A B C D x0 sd- f(x) = ((Acos(x + D)^2 + Bcos(x + D) + C - x0)/sd)^2
-
SOEDINGFUNC w1 mean1 sd1 w2 mean2 sd2- f(x) = ln( w1Gauss(x,mean1,sd1) + w2Gauss(x,mean2,sd2) ) - ln( w2*Gauss(x,mean2,sd2) )
where Gauss(x,mean,sd) is the value of the normal distribution at x, given the mean and standard deviation
-
TOPOUT weight x0 limitHarmonic near x0, flat past limit
- f(x) = weight * limit^2 * ( 1 - e^( -(x-x0)^2/limit^2 ) )
-
ETABLE min max [many numbers]Defines a function via values ranging from min to max inclusive, spaced out by 0.1. Linear interpolation for intermediate values.
USOG num_gaussians mean1 sd1 mean2 sd2...
Defines an unweighted sum of a given number of Gaussians
-
SOG num_gaussians mean1 sd1 weight1 mean2 sd2 weight2...
Defines a weighted sum of a given number of Gaussians
Function types are all implemented as subclasses of the core::scoring::constraints::Func class.
Sample Files
AtomPair CZ 20 CA 6 GAUSSIANFUNC 5.54 2.0 TAG
AtomPair CZ 20 CA 54 GAUSSIANFUNC 5.27 2.0 TAG
AtomPair CZ 20 CA 50 GAUSSIANFUNC 5.26 2.0 TAG
AtomPair CZ 20 CA 10 GAUSSIANFUNC 4.81 2.0 STILL_TAG
AtomPair CZ 20 CA 41 GAUSSIANFUNC 9.90 2.0 I_AM_ANNOYED_BY_THIS_TAG_FIELDAtomPair SG 5 V1 32 HARMONIC 0.0 0.2
Angle CB 5 SG 5 ZN 32 HARMONIC 1.95 0.35
AtomPair SG 8 V2 32 HARMONIC 0.0 0.2
Angle CB 8 SG 8 ZN 32 HARMONIC 1.95 0.35
AtomPair NE2 13 V3 32 HARMONIC 0.0 0.2
Angle CD2 13 NE2 13 ZN 32 HARMONIC 2.09 0.35
Dihedral CG 13 CD2 13 NE2 13 ZN 32 CIRCULARHARMONIC 3.14 0.35
AtomPair SG 18 V4 32 HARMONIC 0.0 0.2
Angle CB 18 SG 18 ZN 32 HARMONIC 1.95 0.35#discourages residue 16, chain B, from interacting with chain A; encourages residue 47
SiteConstraint CA 16B A SIGMOID 5.0 -2.0
SiteConstraint CA 47B A SIGMOID 5.0 2.0AtomPair H 6 HA 134 BOUNDED 1.80 5.25 0.50 NOE ;dist 5.000 1.800
AtomPair HA 7 HA 132 HARMONIC 1.2 0.2AtomPair CB 31 CB 43 SPLINE EPR_DISTANCE 6.0 4.0 0.5
AtomPair CB 29 CB 62 SPLINE EPR_DISTANCE 15.0 4.0 0.5Code for Reading Constraints
There are different sets of functions for reading full-atom and non-full-atom constraints, as indicated by the respective functions listed below. The only current difference between the functions are which command line arguments are read. The values of the arguments are processed identically.
To use constraints, both the scoring function and pose objects should be updated. The functions for adding constraints to the scoring function are:
- core::scoring::constraints::add_fa_constraints_from_cmdline_to_scorefxn()
- core::scoring::constraints::add_constraints_from_cmdline_to_scorefxn()
Currently these functions only set the weights of the atom_pair_constraint, angle_constraint, and dihedral_constraint score function terms to the value of either the -constraints:cst_fa_weight or -constraints:cst_weight command line argument.
The functions for adding constraints to the pose object are:
- core::scoring::constraints::add_fa_constraints_from_cmdline_to_pose()
- core::scoring::constraints::add_constraints_from_cmdline_to_pose()
These functons read a random constraint file from the list defined by either the -constraints:cst_fa_file or -constraints:cst_file argument.
There are also convenience functions for doing both at once:
- core::scoring::constraints::add_fa_constraints_from_cmdline()
- core::scoring::constraints::add_constraints_from_cmdline()
Other Constraint Types
These constraint types cannot currently be specified in a file. They need to have read_def methods implemented and be added to the ConstraintFactory constructor.
- BackboneStub
- Constant
- ResidueType
- Rotamer
Other Function Types
These function types cannot currently be specified in a file. They need to have read_data methods implemented and be added to the FuncFactory constructor.
- ChainbreakDist
- CharmmPeriodic
- CircularPower
- Etable
- CB_Angle
- CBSG_Dihedral
- SG_Dist
- SGSG_Dihedral
- LK_Sigmoidal
See Also
- File types list: List of file types used in Rosetta
- Rosetta Basics: The Rosetta Basics home page
- Match constraints files: Rosetta matching constraints file format (specifically for the match application)
- Options overview: Overview of Rosetta command line options
- Glossary: Brief definitions of Rosetta terms
- RosettaEncyclopedia: Detailed descriptions of additional concepts in Rosetta.
- Rosetta overview: Overview of major concepts in Rosetta
- Application Documentation: Links to documentation for a variety of Rosetta applications