
Under the description for the mover RepackMinimize:(https://www.rosettacommons.org/docs/latest/scripting_documentation/RosettaScripts/Movers/movers_pages/RepackMinimizeMover)
it says "interface_cutoff_distance: Residues farther away from the interface than this cutoff will not be designed or minimized."
However, what is "the interface"? Is it:
1). Within the context of a project where I have two protein chains next to each other (chain A & chain B), is Rosetta somehow pre-determining a set of residues on chain A & B that it calls "the interface",
OR
2). by setting the interface_cutoff_distance to some value (let's say 8 angstroms), am I the one that's telling Rosetta, "hey consider all pairs of residues on chain A & B respectively that are within 8 angstroms of each other, to be the interface"?
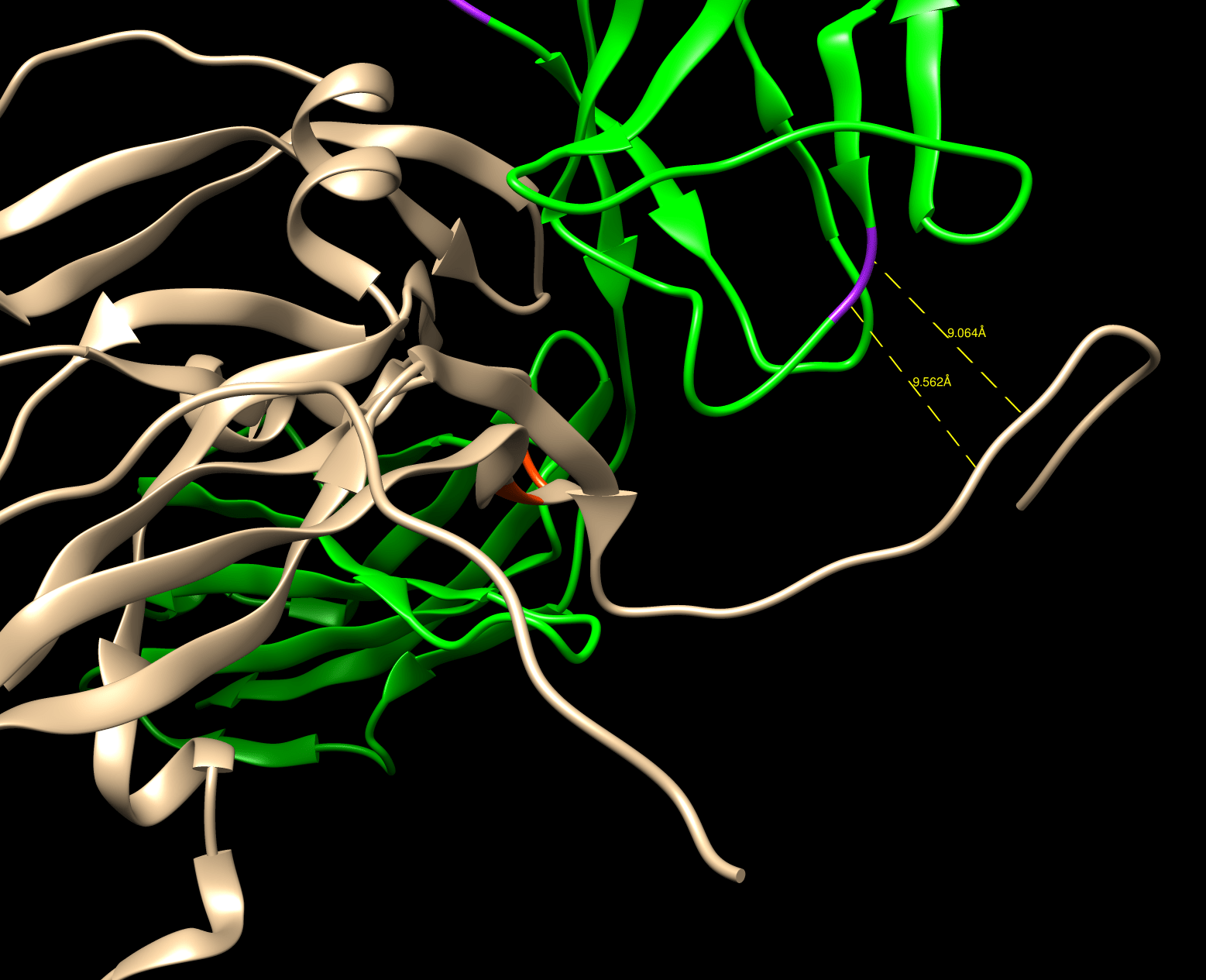
My confusion is worsened by the fact that neither explanation above seems to explain the attached picture.
The attached picture is showing my input structure before running the script below, which uses RepackMinimize as the main mover: chain A is brown, chain B is green, and purples shows some of the residues that were re-designed. According to the RepackMinimizeMover link above, RepackMinimize has a default interface_cutoff_distance of 8.0 angstroms. The picture shows the closest distance between the re-designed residues and chain A to be about 9 angstroms. So why did these residues get re-designed?
For reference, here is the full script I used:
<ROSETTASCRIPTS>
<TASKOPERATIONS> You can control which parts you want to change from here
</TASKOPERATIONS>
<FILTERS> This part enables to output useful values for post-processing
<Ddg name="ddG" scorefxn="talaris2014" threshold="-15" repeats="2"/> binding energy calculation; an average of two repeats is computed for better numerical accuracy
<Sasa name="sasa" threshold="100"/> Buried surface area upon complex formation
<Rmsd name="rmsd" confidence="0"/> confidence=0 means that the filter will be evaluated but not used as an acceptance criterion
<CompoundStatement name="ddg_sasa"> combine filters into a single logical statement
<AND filter_name="ddG"/>
<AND filter_name="sasa"/>
</CompoundStatement>
</FILTERS>
<MOVERS>
<Docking name="docking" score_high="soft_rep" fullatom="1" local_refine="1"/> Invokes RosettaDock local-refinement (in full-atom) with a soft potential
<BackrubDD name="backrub" partner1="0" partner2="1" interface_distance_cutoff="8.0" moves="1000" sc_move_probability="0.25" scorefxn="talaris2014" small_move_probability="0.15" bbg_move_probability="0.25"/> perturb the backbone of chain2. Change moves to 1000 for real application
<MutateResidue name="D63A" target="63A" new_res="ALA"/>
<MutateResidue name="E64A" target="64A" new_res="ALA"/>
<MutateResidue name="E67A" target="67A" new_res="ALA"/>
<RepackMinimize name="des1" scorefxn_repack="soft_rep" scorefxn_minimize="soft_rep" minimize_bb="0" minimize_rb="1"/>
<RepackMinimize name="des2" scorefxn_repack="talaris2014" scorefxn_minimize="talaris2014" minimize_bb="0" minimize_rb="1"/> Design and minimization at the interface
<RepackMinimize name="des3" minimize_bb="1"/>
<ParsedProtocol name="design">
<Add mover_name="des1"/>
<Add mover_name="des2"/>
<Add mover_name="des3"/>
<Add mover_name="backrub"/>
<Add mover_name="des3" filter_name="ddg_sasa"/>
</ParsedProtocol>
<GenericMonteCarlo name="iterate" scorefxn_name="talaris2014" mover_name="design" trials="5"/> Iterate through design to find best solution. change trial numbers to higher for real application.
</MOVERS>
<PROTOCOLS>
<Add mover="docking"/>
<Add mover="D63A"/>
<Add mover="E64A"/>
<Add mover="E67A"/>
<Add mover="iterate"/>
<Add filter="ddG"/>
<Add filter="sasa"/>
<Add filter="rmsd"/>
</PROTOCOLS>
</ROSETTASCRIPTS>
| Attachment | Size |
|---|---|
| 223.19 KB |
{kind=link}