
Dear all,
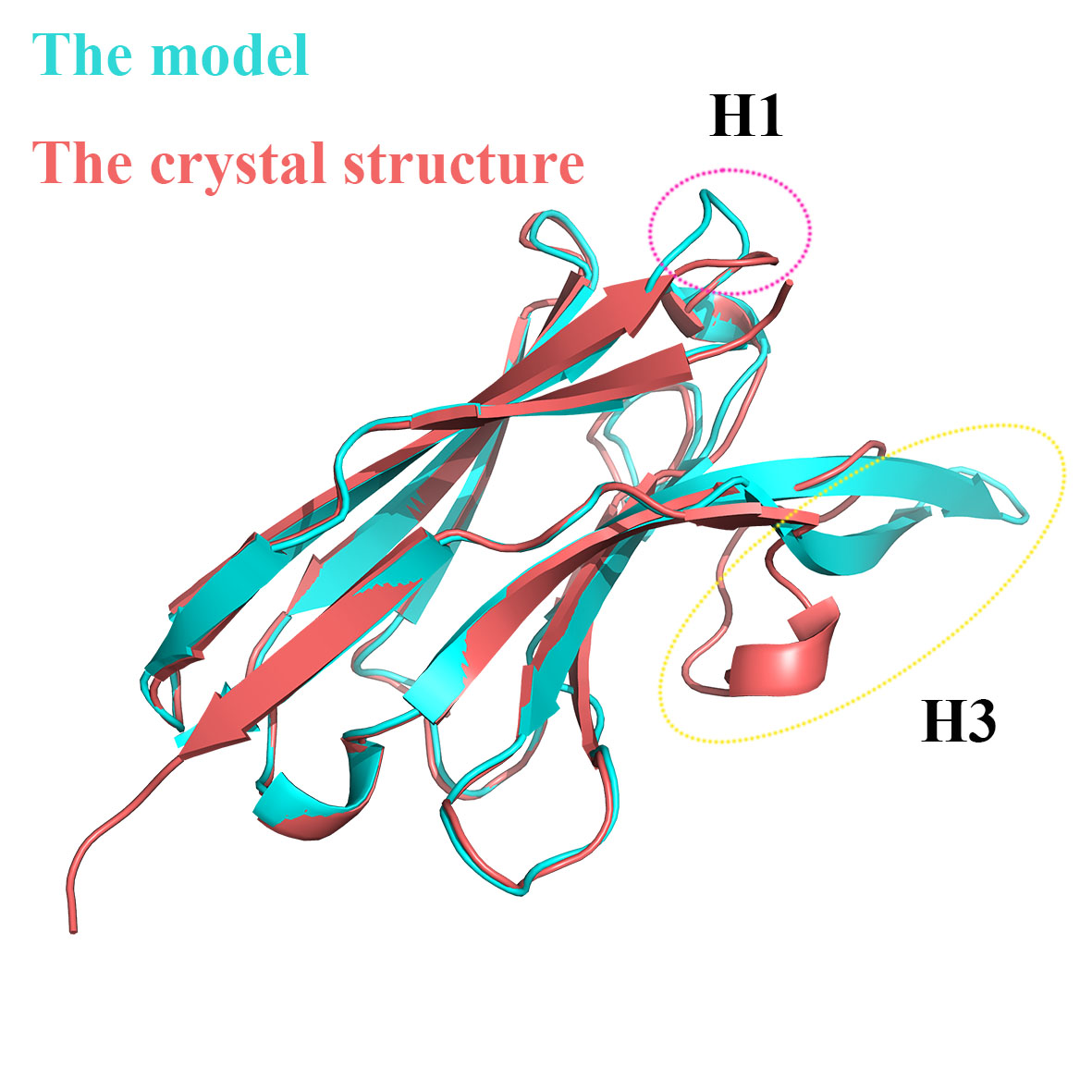
I modeled a camelid antibody structure with "antibody.linuxgcc.release" from its fasta sequence. However, after aligning the modeled structure to its crystal structure, I found that there are obvious difference between the H1 region of these two structures (Please see the attached figure). I wish to use this model to predict the structure of the antibody in complex with an antigen with "docking_protocol.linuxgcc.release". I guess this difference may affect the accuracy of the predicted complex structure.
The H3 region structure can be optimized with "antibody_H3. linuxgcc.release", but is there a way to optimize the H1 and H2 regions? Can "relax.linuxgcc.release" be used for H1 and H2 optimization? If it can, how to set the options?
Thank you in advance!
| Attachment | Size |
|---|---|
| 210.63 KB |
{kind=link}
H1 templates are identified from the input sequence through a BLAST of the template database. Is it possible that other top hits in the BLAST result have a better conformational match to the crystal structure? (Or, in cases where there isn't experimental structural data, do you have some other knowledge of the conformation, like a strong prediction of the conformation cluster that is not being picked up by BLAST?) I'd recommend you look through the blast results in grafting/h1.align (one of the outputs from the antibody application) and then use the command-line options to specify the template for H1 if you find one with a better match.
That said, if you have the crystal structure you should use this for your docking study instead of a model. Docking results are almost always better when starting from a crystal structure.
Relax is run by default as part of the antibody modeling protocols and does work on the full protein. However, I'm not sure that it provides the type of conformational search you would need to accomplish a loop reorienation like you're looking for. You could think about using the loop building algorithms on H1 but there are several reasons not to pursue this: 1) there is a crystal structure of the antibody already; 2) sometimes the electron density in crystal structures is improperly modeled. Most antibody CDRs fall into a known cluster and many singletons are the result of mistakenly modeled density in the crystal structure. You can use the identify_cdr_clusters application to evaluate the conformational cluster of the crystal structure and models. 3) There is substantial precedent that grafting processes for antibody modeling provide higher quality models than does loop building alone. Except that you already have the crystal structure, there is no reason to expect that the model is wrong (unless there is something unique about your sequence that precludes it from falling into a known cluster, which is very rare.)