
Hello, i am trying to relax an enzyme with a co-factor (nadp) but whenever I do, the resulting structure looks wrong.
I use the NADP structure from pubchem as a starting point, then molfile2params structure and copy paste the returned LG_0001.pdb to my protein.
i then run the following code:
init("-default_max_cycles 200 -ignore_unrecognized_res 1 -ex1 -ex2aro -ignore_zero_occupancy false -fa_max_dis 9")
#read paramsfile and pdb file and create initial pose
ligand_params = [LG1.params]
pose = pyrosetta.rosetta.core.pose.Pose()
if len(ligand_params) != 0 and ligand_params[0] != "":
ligand_params = pyrosetta.Vector1(ligand_params)
res_set = pose.conformation().modifiable_residue_type_set_for_conf()
res_set.read_files_for_base_residue_types(ligand_params)
pose.conformation().reset_residue_type_set_for_conf(res_set)
pyrosetta.io.pose_from_file(pose, "pathtopose.pdb")
scorefxn = create_score_function("ref2015_cart")
pose_relax = pose.clone()
scorefxn = create_score_function("ref2015_cart")
relax = pyrosetta.rosetta.protocols.relax.FastRelax()
relax.set_scorefxn(scorefxn)
relax.cartesian(True)
relax.apply(pose_relax)
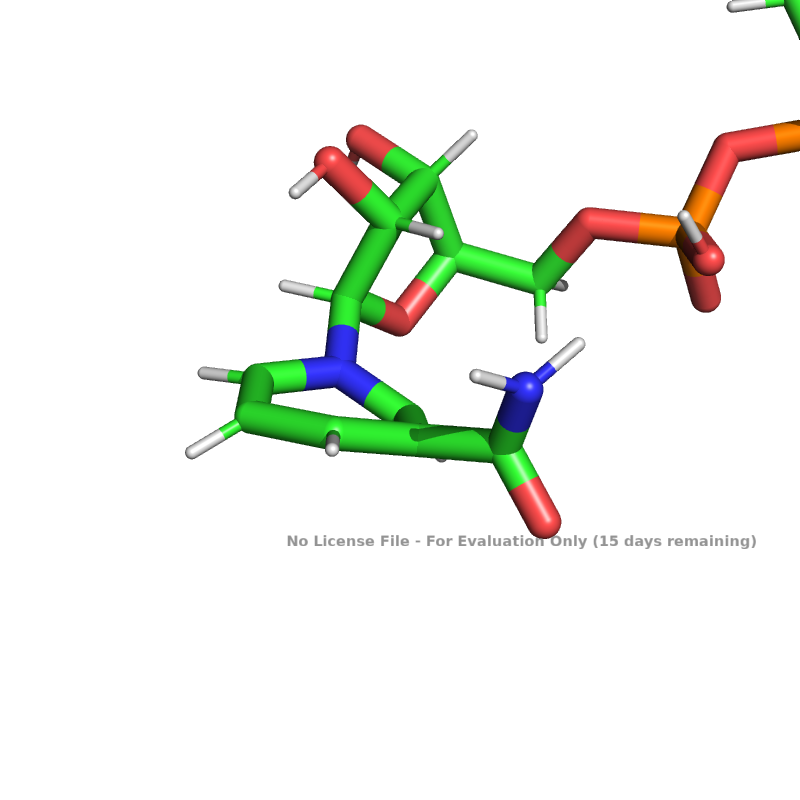
however when i run this and dump the resulting pdb file, what i end up with looks like this:
https://imgur.com/a/riWsjOL (sry having issues displaying the image)
Does anyone know what causes this?
thanks in advance,
David

| Attachment | Size |
|---|---|
| 97.28 KB |
{kind=link}
Category:
Post Situation:
You're doing a Cartesian relax, which allows more internal flexibility of the ligand. You've enabled the cart_bonded terms with the ref2015_cart score function, which is good and will keep bond lengths and angles from varying too much. However, I don't know how good the internal improper dihedral restraints are for ligands. Not having those on fully might cause the issues you see where the ring starts to get distorted.
The molfile_to_params.py script has a `--extra_torsion_output` option, which should output additional cart_bonded parameters for the improper dihedrals of the ligand. You should then be able to pass that to Rosetta with the `-extra_improper_file` parameter, and the cart_bonded term should use those parameters to further constrain the ligand. This may help with the issue you're seeing.