
Dear All,
Any one knows why the Rosie Server for protein docking (docking2) automatically gave two plots that seem not consistent with each other on the x-axis which is the "RMSD". Or me please give some suggestion if I understand something wrong. Please see the attached figure.
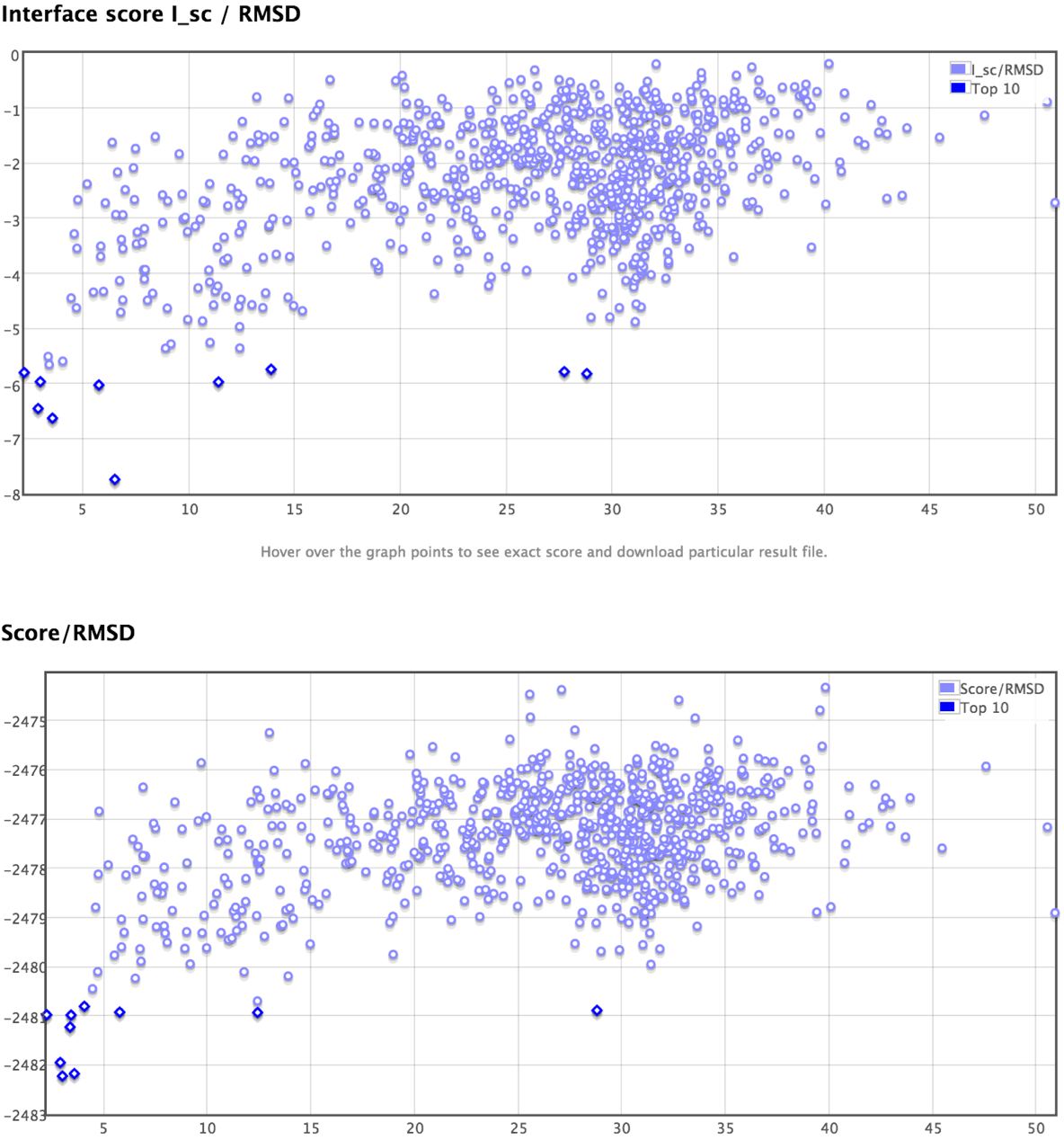
I know from the Rosie website that the clustering of lowest energy poses like a funnel shape is a gold standard for success of identifying a potential binding pocket. But not sure why the top 20 low energy poses show up differently along the x-axis in the two plots, which kind of limits my confidence in the usage of these results...). Thanks for any comments.
Best,
Conan
Rice University
| Attachment | Size |
|---|---|
| 175.11 KB |
{kind=link}
Category:
Post Situation:
Conan, please see below reply to your question from one of our lab members.
------------------------
Hi Conan,
The upper panel is the Interface score plotted over the RMSD whereas the lower panel is the total score over the RMSD. The total score gives you the overall "energy" of the complex, while the interface score is the total score of the complex minus the total score of each partner in isolation. It's kind of like the energy that is contained in the interface. The interface score is typically a better indicator of a better docking complex. You can also find more information here:
https://www.rosettacommons.org/docs/latest/docking-protocol.html#Tips
The x-axis is the RMSD, which doesn't change, just the score of each model is different in the two different plots.
As for clustering of the models, the top10 models cluster into three clusters when you look at the interface score: 0-7A with 6 models, 10-15A with 2 models and ~30A with 2 models. A higher number of models in a cluster points to a higher confidence. This means that the models in the first cluster are likely the closer to the native, which in fact, they are.
Hope this answers your question.
Best,
Julia
Thank you so much, Julia and your colleague. I think understand the explanations now.
However, It's just that the top 10 models comparing the top and bottom graphs somehow reside differently along the x-axis (especially the interface score plot the actually individual point not all correspond to their RMSD correctly), which can be a output plot error every single case I tried with the Rosie Docking2 server online. If you look at the originally attached example closely comparing the two graphs, you might find out... which will be something the server can fix I guess unless I am totally wrong about the observation.
Thanks again.
Conan
The top 10 in each case is different. In the top panel it's the top ten by interface score, and in the bottom it's the top 10 by total score - the two sets of 10 are different structures. On the results page you should be able to mouse over the points, and it will tell you which structure each point corresponds to. If you do so, you'll see that the two sets of 10 are different structures.
Now I get it... Thanks a lot!
I think there is indeed a problem with the I_sc/RMSD plots.
Look at http://rosie.rosettacommons.org/docking2/viewjob/16276 for example:
The left-most dark blue circle has (RMSD,I_sc) near (1,-7.7),
but if you hover over this circle, it says it is for the proteins_0201.pdb file
with (RMSD,I_sc)=(9.26,-6.962), numbers that agree with the spreadsheet further down.
The right-most dark blue circle has (RMSD,I_sc) near (23,-6.7),
but if you hover over this circle, it says it is for the proteins_0244.pdb file
with (RMSD,I_sc)=(12.965,-5.324), numbers that agree with the spreadsheet further down.
If you sort the spreadsheet on the I_sc column, the proteins_*.pdb files with the 10 lowest I_sc scores:
0042, 0210, 0949, 0201, 0122, 0002, 0672, 0734, 0043, 0482
should match the files for Models-1 to 10 in the top-10 rainbow-colored 3D images (they do)
as well as the dark blue circles on the I_sc/RMSD plot (they don't).
Instead, the dark blue circles on the I_sc/RMSD plot from bottom to top are listed as:
0042, 0201, 0002, 0210, 0949, 0244, 0734, 0234, 0462, 0043.
Not only has the order changed, but different files are listed!
This 2nd list matches the files with the 10 lowest total_scores when the spreadsheet is sorted by total_score
as well as the files listed for the dark blue circles on the Score/RMSD plot from bottom to top.
I have seen many errors like this on I_sc/RMSD plots from my own docking2 jobs
but never on the same jobs' Score/RMSD plots.
It seems like https://www.rosettacommons.org/node/3911 discusses the same kind of error.
If possible, please fix these errors so future users won't be confused by the I_sc/RMSD plot labels.
Thanks!
The window that pops up when you hover the cursor over a point on an I_sc/RMSD or Score/RMSD plot could also be improved.
I suggest adding a blank line at the top of each window so that the cursor doesn't cover the pdb file name.
Thanks again!