
I am now working on a protein with multiple chains, and the total amino acids exceeds 1000. So I need to submit each chain one by one, get the individual 3-mer and 9-mer fragments, then concatenate them.
http://old.robetta.org/fragmentsubmit.jsp
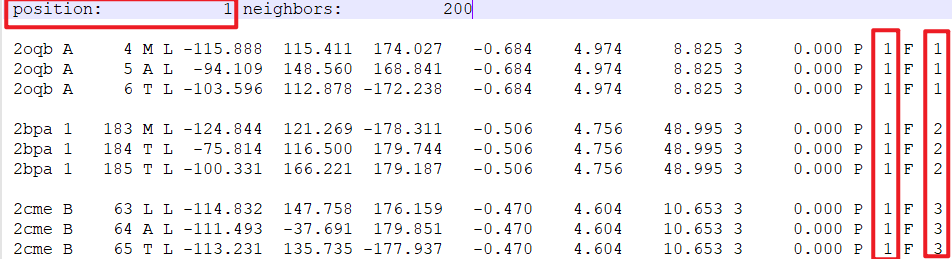
The attachment is the 3-mer file. After concatenating them, do I only need to adjust these three places?
I will then use the 3-mer and 9-mer for the RosettaCM.
By the way, I plan to use RosettaCM to build a structure of 16,000 residues, which is an asymmetric unit of an adenovirus. Is it possible?
Any feedback is welcomed! Thank you!
| Attachment | Size |
|---|---|
| 33.86 KB |
{kind=link}
Category:
Post Situation:
I believe all you need to do is make sure the 'position' is reset to be the next number after the end of the previous fragment
so with 99 positions in the first fragment file, and the next one starting at 1, you would have to edit it to be position 100
16,000 residues is a lot, I believe the most i have done is 12,000.
some things to keep in mind:
#1 ~6,000 residues takes about 24 hours, i imagine this will take ~3 days.
#2 This will take a lot of memory, I'm not sure exactly but it's possible it could take up to (or even more) than 30G
#3 If your sequence isn't properly aligned in the template pdb file to the fasta file, it will do a SmithWaterman alignment to try and align it, this will also take a lot of memory at 16,000 residues
I would suggest doing a fastrelax first with your input structure to test the waters for the memory requirements of packing with this many residues.
Also I wouldn't expect with 16,000 residues for the sampling of loops to be 'great', in the past i've used divide and conquor approaches for things like this were i have done multiple smaller ~1-3,000 residues CM jobs at a time, and then combined them all at the end so that everything got sampled a little more.
Dear Danpf,
Thank you! This is very good advice, especially the memory estimation! Do you also have an estimation for the cores needed?
I think I may eventually model smaller units then "combined them all at the end" as you said. Can I ask which protocol do you use to "combined them all at the end"?
Best regards
Cheng
> estimation for the cores needed
Rosetta is not multithreaded, so 1 core is fine.
I would run CM ~30-50 times and take the best scoring result, but I wouldn't expect much to change between them in all honesty.
> Can I ask which protocol do you use to "combined them all at the end"?
I would run CM on smaller complexes at first, then at the end I would use pymol or a text editor to add all the pdbs together into one, and then do another 'final' CM job (the same as the one you're proposing to do now). This has the benefit of additional sampling to each peptide, but might not be necessary depending on the quality of the template you're using.
Thank you Danpf!
I will see what it looks like when combining the small units into one. I feel like the coordinates will deviate after RosettaCM for each small unit. Hope it won't.
Also, you mentioned Rosetta is only for 1 core. Can I clarify, do you mean RosettaCM is only for one core, or all the Rosetta protocols are for one core? We can run Rosetta on our High-performance computers using mpi with multiple cores, is it necessary?
Almost all rosetta protocols are meant to be run on 1 core. mpi should work though!, although I've never used it (i know lots of people do) -- using mpi cuts down on memory usage which is very nice.
The point i was trying to make is -- running a non mpi rosettaCM rosetta_scripts with 20 cores will not be faster than running it on 1 core.