
Dear all,
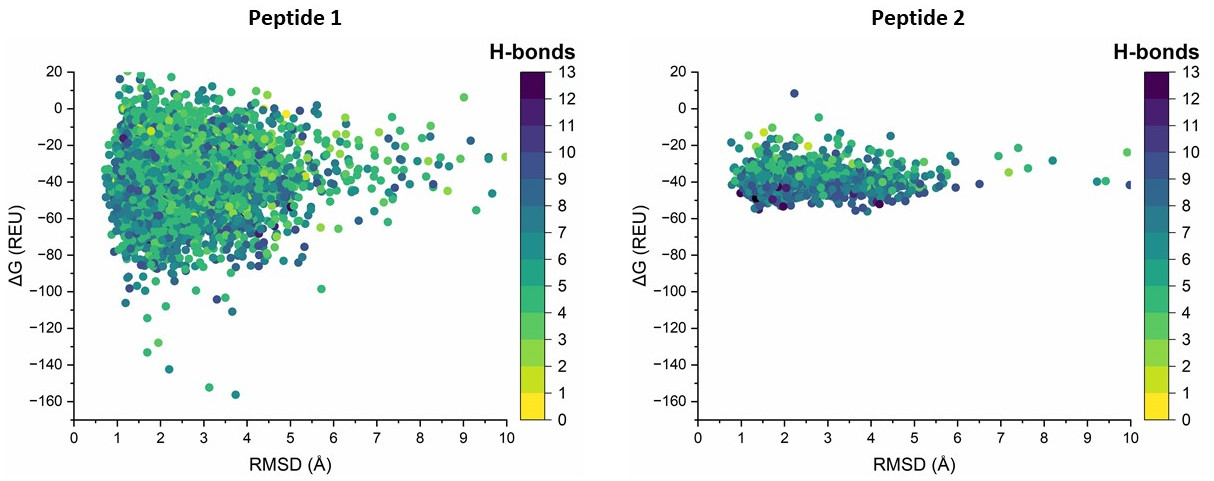
I am designing some peptides with the anchor_design approach. I am using the dG_separated values to compare them. For this, I included an InterfaceAnalyzer mover in my anchor_design xml script as follows:
<InterfaceAnalyzerMover name="interface_analyzer" scorefxn="r15" pack_separated="true" pack_input="true" packstat="true" interface_sc="true" interface="A,B" />
Attached is a plot of the sampling of two peptides. According to the thread https://www.rosettacommons.org/node/9822 the values may be too low. The thread suggested to use -extrachi_cutoff 0 but when I run the standalone application with one of the decoys of peptide 1 as:
InterfaceAnalyzer.linuxgccrelease -database $ROSETTA_DB -in:file:s complex.pdb -interface A_B -compute_packstat true -pack_separated true -add_regular_scores_to_scorefile true -out:file:score_only IA_score.sc -extrachi_cutoff 0
the value is even more negative than the one obtained with the anchor_design script.
My question is, do these values make sense for the comparison between the peptides?
Thanks in advance.
| Attachment | Size |
|---|---|
| 101.43 KB |
{kind=link}